
臨床ラベルの翻訳と規制要件を管理する方法に関する重要なヒント
April 10, 2022 (2 分で読めます)

カテゴリー| 治験関連サービス
新しいEU臨床試験規則(CTR)は、治験管理をシンプル化し、製薬企業が欧州で治験を実施しやすい環境を構築することを目指して制定されました。CTRが法的拘束力を持つようになり、薬事、ラベリング、Qualified Person(QP)要件が統一された今、各治験依頼者はその内容を十分に把握しなければなりません。CTRが製薬業界に与える影響について、規制遵守を確保する方法も含めて解説いたします。規制遵守には、経験豊富な人材と変化への対応力が必要です。そして、治験に関する高い専門知識を備え、世界各地に拠点を有し、実績ある技術、そしてリスク低減戦略を提供できるパートナーとの提携が必要かもしれません。
制定までに活発な議論がなされましたが、CTRでは、治験薬への電子ラベル(eLabel)の使用は認められていません。使用期限は、従来のブックレットラベルにより、治験薬の直接包装および二次包装の双方に印字する必要があります。
CTRのラベリング要件は、「使用期間」の表示、すなわち治験薬(Investigational Medicinal Product:IMP)や治験使用薬(Auxiliary Medicinal Product:AMP)の使用期限に焦点を当てています。使用期限は、各薬剤の直接/一次包装および二次包装に表示しなければなりません1。

これらの新しい要件への対応にあたって、製造業者からは複数の領域に関する懸念が指摘されています。
臨床試験で使用される治験薬や治験使用薬の使用期限(使用期間)は、例外なく直接包装に表示しなければならないことになりました2 。これは、EU臨床試験規則536/2014の付属書VIの要求事項です。この結果、有効期間の延長が非常に難しくなり、電子システムの導入が不可能となっています。
上記リストの最後の項目、電子ラベルには様々な種類があり、クラウドにより速やかに更新可能なものもあります。バーコード、クイックレスポンス(QR)コード、無線自動識別(RFID)タグ、包装への貼付・変更が可能な電子ペーパーなどがその例です(図1)。
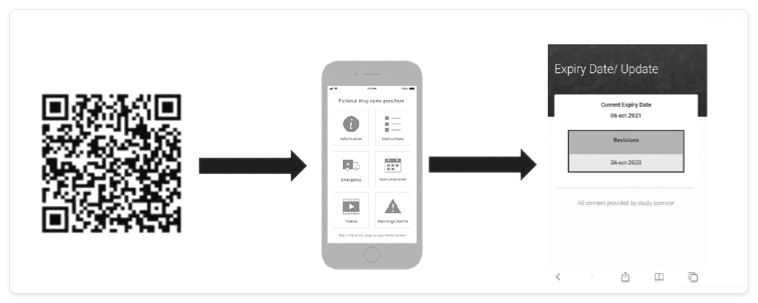 Figure 1. Potential solutions
Figure 1. Potential solutions
電子ラベルの基本機能とそのメリットは以下のとおりです。

 Figure 2. Electronic labeling: functionality and benefits
Figure 2. Electronic labeling: functionality and benefits
世界各国のバイオ医薬品R&D企業と提携し、さまざまな領域の改善に取り組む組織TransCelerateの調査では、電子ラベルは患者さん、治験実施医療機関、規制当局、治験依頼者に次のようなメリットをもたらすことが明らかとなっています3。
これまでに行われた電子ラベルの普及活動の例としては、大手製薬企業7社が2020年に着手した取り組みがあります4 。Alliance to Modernize Prescribing Information(添付文書改革連合)という名称で活動するこのグループは、紙ベースのラベリングは時代遅れであり、患者さんへの安全性情報提供のための技術フローに適合していないと指摘しています。また、平均年5回必要となる承認済みラベルの改訂は、時間のかかる作業であり、タイムラグが生じる可能性があることから、患者さんの安全性に対するリスクとなることを指摘しているグループもあります5 。いずれも妥当な指摘であり、臨床試験で使用する治験薬についても電子ラベルを導入すべきときが来ているのかもしれません。これを実現する有望なアプローチとして、法令で要求されているブックレットラベルと平行して、薬剤が使用されるすべての国の言語での患者向け情報の提供など小規模での電子ソリューションを導入する方法があります。患者さんの安全性の確保あるいは向上を実現できることが確認されれば、規制当局の信頼を得るために必要なproof of concept(POC)取得につながる可能性があります。
ただし、新しい選択肢を検討する際には、患者さんの安全性確保が常に最優先事項であることを忘れてはなりません。
External partners can help fulfill sponsor needs for labeling compliance support, whether for individual elements to supplement a big pharma company’s own capacity or for a full range of end-to-end packaging and clinical trial services for an emerging biotech. Sponsors looking to partner on labeling advances should evaluate several qualities in potential partners:
新しいラベリング要件の遵守というハードルを乗り越えられれば、EU加盟国間・加盟国内の連携、情報共有、意思決定の強化など、新しいCTRが意図するメリットを享受することができるでしょう。