
医薬品開発における5つのフェーズ(段階)について
Category | CDMO Services
医薬品開発は紛れもなく複雑である。
分子の発見からFDAの審査、安全性のモニタリング、商品化まで、物事がうまくいく機会もあれば、うまくいかない機会もたくさんある。2022年にFDAが承認した新薬は37品目であった1。実際、新薬の成功確率は全体でわずか12%である2。
新薬が「成功」とみなされるには、5つの段階を経る必要がある: 1)発見と開発、2)前臨床研究、3)臨床研究、4)FDA審査、5)安全性モニタリング。FDAの審査、そして5)安全性モニタリングである。
以下、各段階について詳しく説明する。

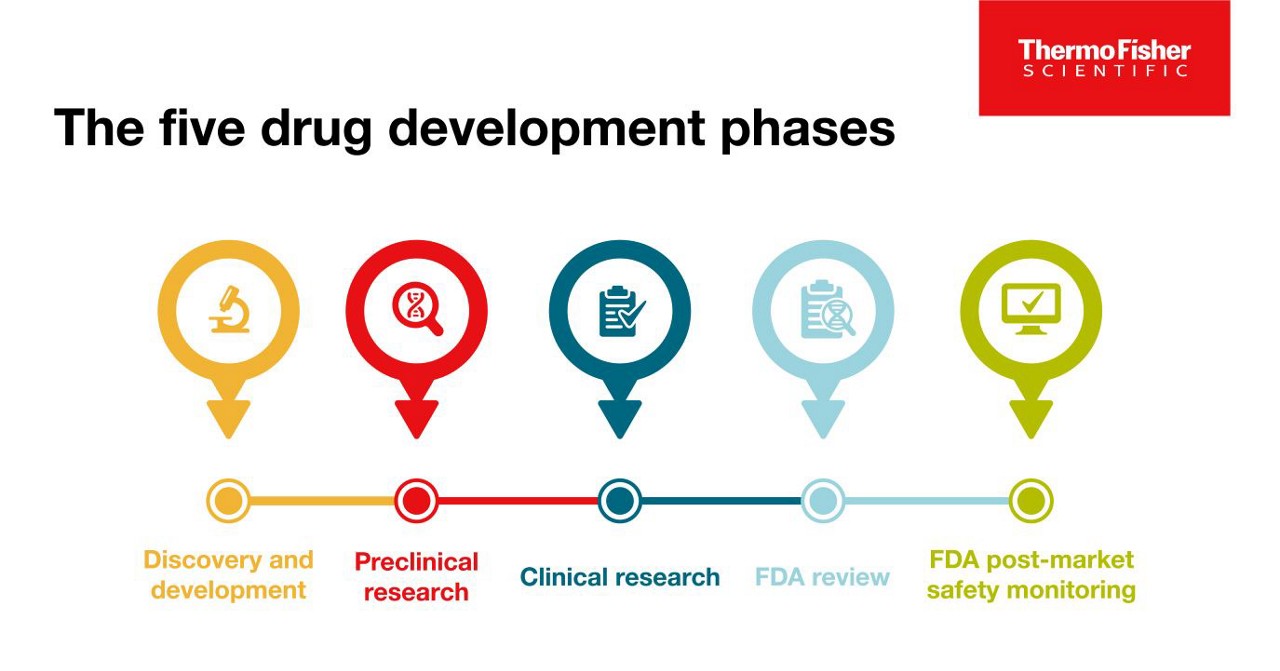
第一段階:ディスカバリーと開発
医薬品の開発・製造が行われるずっと前に、研究者は特定の分子(通常はDNA配列、RNA分子、タンパク質、代謝産物など)を「発見」しなければならない。発見後、研究者は標的分子と相互作用し、薬剤候補となる可能性のある化合物を探索することができる。通常、さまざまな化合物が候補として特定されるため、研究者はそれぞれの化合物について一連の実験を行い、最終的な最も効果的な薬物としての性能と実行可能性を評価しなければならない。一般的には、吸収、投与、副作用、相互作用の可能性などの要素を評価する。これらの実験が最終的に終了し、最も有望な化合物が単離されると、前臨床研究の段階に入ることができる。
「前臨床研究の目的は、化合物が重大な害をもたらす可能性があるかどうかを評価することにある」
第2段階:前臨床研究
化合物をヒトで試験する前に、研究者はin vitro(試験管内)またはin vivo(動物)のいずれかで前臨床研究を実施しなければならない。前臨床研究の主な目的は、化合物が重篤な害を引き起こす可能性があるかどうかを評価することである。さらに、候補化合物は薬力学と薬物動態学の試験を受け、薬物が身体に与える作用と身体が薬物に与える作用を調べる。前臨床研究はまた、安定性、生物学的利用能、投与方法などの製剤開発を決定する上で重要な役割を果たします。すべての前臨床研究は、データの質、完全性、信頼性の基準を定めたFDAのGLP(Good Laboratory Practice)規制を遵守しなければなりません。医薬品が前臨床研究を成功裏に通過した後に、ヒトで試験する準備が整うことになります。
第3段階 臨床研究
臨床試験による臨床研究は、医薬品開発の次のステップであり、化合物の安全性と有効性をヒトで試験する役割を果たす。臨床研究は通常、第I相、第II相、第III相、第IV相の4段階に分けられる。FDAによれば、第I相には20〜100人の健常人ボランティアまたは疾患や病態を有する人が、第II相には最大数百人の疾患や病態を有する人が、第III相には300〜3,000人の疾患や病態を有する人が、第IV相には数千人の疾患や病態を有する人が参加する。臨床試験で安全性と有効性が証明されれば、その化合物はFDAの審査に進むことができる。先に述べたように、臨床試験に参加した新薬のうち、最終的にFDAの承認を受けるのはごく一部であることに注意することが重要である。有効性の欠如や安全性の問題などが、この低い成功率の原因となっていることが多い。
「その医薬品が意図された用途に対して安全かつ有効であると判断された場合、FDAはその医薬品を米国内で製造、販売、流通させる認可を与える」
第4段階 FDA審査
この審査では、医師、化学者、統計学者、微生物学者、薬理学者などの専門家チームが、臨床試験から得られた化合物の安全性と有効性に関する知見を検討する。バイオテクノロジー企業や製薬企業がFDAの審査を受けようとする場合、医薬品の場合は新薬承認申請書(NDA)を、生物製剤の場合は生物製剤承認申請書(BLA)を提出しなければならない。その後、FDAは申請を受理し、そのケースを評価する専門家チームを任命しなければならない。チームは共に、患者の転帰、潜在的な副作用、医薬品のリスク・ベネフィット分析などの臨床研究を検討する。その医薬品が意図された用途に対して安全かつ有効であると判断された場合、FDAは米国内での製造、販売、流通の承認を与える。
第5段階 FDAによる市販後の安全性モニタリング
臨床研究は、比較的少数のボランティアにおける医薬品の安全性と有効性を評価するものであるが、承認後に一般集団において新たな懸念が生じる可能性はある。そこで、FDAの市販後安全性監視(市販後サーベイランス)の出番となる。FDAには、MedWatchやMedSunなど、市販後の安全性監視を支援するプログラムがいくつかある。MedWatchは、医療従事者や消費者が医療製品の重大な問題を報告できるようにするものであり、MedSunは、医療機器の使用に特に関連する問題を特定、理解、解決するために臨床コミュニティと協力するものである。さらに、FDAは医薬品の製造施設が規制基準に適合していることを確認するために定期的な検査を実施し、バイオテクノロジーや製薬企業が虚偽または誤解を招くような主張をしていないことを確認するために医薬品の広告や表示も監視している。
CDMOのパートナーは、医薬品開発における企業の舵取りをサポートします。
しかし、バイオテクノロジー企業や製薬企業は、医薬品開発の道のりを単独で進む必要はありません。しかし、バイオテクノロジー企業や製薬企業が単独で医薬品開発の道を歩む必要はない。CDMO(開発・製造受託機関)と提携することで、医薬品開発、製造、規制遵守に関する業界の専門知識だけでなく、高度な能力やリソースを利用することができる。創薬・開発、前臨床・臨床研究、安全性監視などの主要機能をCDMOにアウトソーシングすることで、バイオ・製薬企業は規制要件へのコンプライアンスを確保しながら、自社の中核的な強みと優先事項に集中することができる。
Contact us to learn more about what makes a quality CDMO, or download our whitepaper below.
View references

More on the topic

Blog post
CROs vs CMOs, and CDMOs: What’s the difference between the three?
CROs, CMOs, and CDMOs all help biotechnology and pharmaceutical companies with drug development and manufacturing, but what’s the difference between the three?
Read Blog
Blog post
What is a CDMO?
Learn how CDMOs (contract development and manufacturing organizations) work with pharma companies, and the top considerations companies should have when choosing a CDMO partner.